
12.14 Genetic approaches to study development: through the classic to genomic systems analysis
The suggestion that there are several fundamentally different ways in which cell inflation and tissue expansion can be achieved is reinforced by work done on the induction of developmental variants. This classic genetic approach to the study of any pathway depends on:
- identification of variant strains;
- complementation tests to establish functional cistrons;
- comparison of heterokaryon phenotypes to determine dominance;
- determination of epistatic relationships in heterokaryons, to indicate the sequence of gene expression (Moore & Novak Frazer, 2002).
This, of course, now looks like a rather old-fashioned approach, though it’s lost none of its power to dissect an individual pathway. The modern molecular approach is to use genomic analysis tools to analyse and compare the transcriptome (transcribed RNA population) and/or proteome (translated polypeptide population) to give expression profiles of different stages as the organism progresses through its developmental sequence. The classic approach (‘first, find a variant’) immediately focuses attention on a decisive event, though it can then be difficult to identify the network of interactions to which this belongs (the network only begins to become evident as more and more individual decisive events are dissected out, which can take a long time and a lot of work).
In contrast, genome-wide analysis gives you an immediate view of the overall grand network, which comprises many thousands of interactions, and it can then be difficult to bring the focus down to identify the few decisive events that enable neighbouring cells in a hypha to embark on different developmental pathways. The abstract of one paper, which describes gene expression studies during fruit body development in ‘our favourite little mushroom’ Coprinopsis cinerea, concludes with the sentence: ‘…We implicated a wealth of new candidate genes important to early stages of mushroom fruiting development, though their precise molecular functions and biological roles are not yet fully known... (Cheng et al., 2013).’
Finding out what all those genes do can take a long time and a lot of work. We will describe some of this work in this Section, as it applies to the organisms we have already mentioned and give more information about the methods used in Chapter 18. If you are impatient to find out about the methods employed, we recommend you refer to the following volumes in the Methods in Enzymology book series: Weissman et al., 2010 (volume 470); Jameson et al., 2011 (volume 500); DeLong, 2013 (volume 531); Doudna & Sontheimer, 2014 (volume 546).
It is easier for us to begin our summary of genetic aspects of fungal developmental biology with a trip down Memory Lane to the pre-genomics era, while we continue our story about Coprinopsis cinerea. The classic genetic approach, when applied to Coprinopsis cinerea, resulted in the isolation of, among others: stipe elongationless mutants, which were unable to elongate the stem but with normal cap expansion, or cap expansionless mutants unable to expand the cap but with normal stipe elongation, and sporeless mutants in which the fruit bodies were morphologically completely normal except for the total absence of spores (Takemaru & Kamada, 1971, 1972) (Fig. 26).
 |
|---|
| Fig. 26. Developmental mutants of the Ink-cap mushroom, Coprinopsis cinerea. Shown from left to right are A and B the wild type (A mid-development, B fully mature and autolysing), and then C sporeless, D cap expansionless, and E stipe elongationless (immature at top, fully mature and autolysing below). The three mutants are all dominant and segregate in crosses as single genes. All cultures are contained in 9 cm diameter crystallising dishes. Photographs by Dr Junxia Ji. |
The three mutants were all dominant and segregated in crosses as single genes. Evidently, the pathways of cap and stem differentiation can be totally separated, and the sporulation pathway is separate from normal cap development. This leads to the conclusion that assembly of different parts of the same fruit body uses genetically distinct pathways. That conclusion can be generalised to: differentiation of fungal multicellular structures is genetically compartmentalised and normal morphogenesis is made up of several-to-many developmental subroutines.
For example, there may be subroutines for stem, cap, hymenium, hymenophore, etc. The subroutines can be put into operation independently of one another and are under separate genetic control and under separate physiological control. Normal morphogenesis of a specific fungus involves integration of these subroutines to produce the fruit body morphology that characterises that species. In their analysis of complex multicellularity in fungi, Nagy et al. (2018) use a slightly different description, suggesting that gene regulatory circuits that orchestrate fruit body development in fungi evolved in a stepwise manner, building on ancient regulatory modules, but also on co-option of conserved genes coupled with the evolution of new ones.
Another important point is that fungi are very tolerant of developmental imprecision. Providing only that the sporulation pathway is intact, even a grossly abnormal fruit body can produce and distribute spores. Variation in the morphology of fruit bodies of higher fungi has been reported in many species, often appearing to be a strategy for adaptation to environmental stress. Detailed analysis of such developmental plasticity in spontaneous abnormal fruit bodies of Volvariella bombycina has given more evidence for normal morphogenesis being an assemblage of distinct developmental segments (Chiu et al., 1989). Although the fruiting structures observed in this study varied from the normal agaric form to completely abnormal enclosed, puff-ball like structures, they were all able to act as production/dispersal structures for basidiospores (Fig. 27).
Thus, a fruit body may, quite abnormally, bear a functional hymenium on the upper surface of the cap, rather than (or even in addition to) its normal hymenium on the lower surface (Fig. 27). The important point is that although the development is abnormal because a morphogenetic subroutine has been invoked in the wrong place, the result is a functional and recognisable tissue, not a disorganised tumorous growth.
 |
| Fig. 27. Polymorphic fruit bodies of Volvariella bombycina. A is the normal morphology, of a typical agaric mushroom, which is characterised by a well-developed enclosing volva through which the mushroom cap emerges during development (bulbangiocarpic development, see Fig. 12.7); (scale bar = 20 mm) B to G illustrate a few of the polymorphisms which have arisen on otherwise normal cultures, though often seeming to be associated with some environmental stress, such as abnormal temperature and/or desiccation. B is apparently normal, but completely lacks the volva despite this being characteristic of the genus (scale bar = 5 mm). C, a gymnocarpous fruit body (see Fig. 12.3) with a grossly enlarged basal volva (scale bar = 2 mm). D, a fruit body with a sinuous extra hymenium formed by the proliferating margin of the cap curling onto the upper surface of the cap (scale bar = 10 mm); E is a scanning electron microscope (SEM) view of specimen D and shows the well developed gills on the upper surface of the cap (scale bar = 1 mm). F, a fruit body in which hymenium has formed all over the outer surface of the club-shaped fruit body (scale bar = 5 mm); G is an SEM view of the bisected fruit body and shows that the labyrinthine hymenophore covers the entire surface of the club-shaped ‘pileus’ (scale bar = 0.5 mm). This polymorph is called ‘morchelloid’ because of its resemblance to the normal fruit body of the morel (Morchella), which, of course, is a member of the Ascomycota! Photomicrographs by Prof. S.-W. Chiu, Chinese University of Hong Kong. Illustration modified from Chiu et al., 1988. |
This tolerance of imprecision is an important biological attribute of fungal morphogenesis, as it provides the flexibility in expression of developmental subroutines that allows the fruit body to react to adverse conditions and still produce a crop of spores. Even when conditions are adverse to normal development, there is still sufficient flexibility in the developmental programme for the fruit body to fulfil its function (Moore, 2013). Similar developmental defects have been described in Pleurotus (Okuda et al., 2019).
The possibility has been discussed that fungal differentiation pathways exhibit what would be described as ‘fuzzy logic’ in cybernetic programming terms (Moore, 1998b). Instead of viewing fungal cell differentiation as involving individual major (yes/no) ‘decisions’ which switch progress between alternative developmental pathways that lead inevitably to specific combinations of features, this idea suggests that the end point in fungal differentiation depends on the balance of a network of minor ‘approximations’. Fuzzy logic is an extension of conventional (Boolean) logic that can handle the concept of partial truth, that is truth-values between ‘completely true’ and ‘completely false’. It is the logic underlying modes of decision-making that are approximate rather than exact, being able to handle uncertainty and vagueness and has been applied to a wide variety of problems.
The importance of fuzzy logic derives from the fact that the theory provides a mathematical basis for understanding how decision-making seems to operate generally in biological systems, which are not always ‘all-or-none’ decisions. In the case of fruit body structure, the outcome is that fungal cells are allowed the flexibility to assume a differentiation state even when all conditions of that state have not yet been met. In other words, developmental decisions between pathways of differentiation are able to cope with a degree of uncertainty. So the conclusion is that fungal differentiation pathways must be based on application of rules that allow considerable latitude in expression (fuzzy constraints), which in the ultimate can lead to highly polymorphic, yet functional, fruit bodies.
One consequence of this line of argument is that patterning genes may not be necessary to develop the patterns of a mushroom fruit body. Rather, as we have described, the patterns may arise from the regular application of simple, and fuzzy, developmental rules and basic metabolic regulation combined with physical forces (stretching, inflation, expansion). A specific gill pattern in an agaric, say, may not require a panel of regulatory genes that specify ‘x number of gills per unit volume of cap’ because that pattern ‘invariably’ results from the application of general rules about the local circumstances within the tissue that allow gills to be made and the subsequent set of circumstances that drive fruit body maturation.
The first complete genome sequence of any eukaryote to be released, in 1996, was that of the budding yeast Saccharomyces cerevisiae. It was the work of a worldwide team of hundreds of researchers (Goffeau et al., 1996). In the time since then, the yeast genome has been intensively studied (Cuomo & Birren, 2010) exemplifying numerous approaches that have been followed with other organisms. An updated reference genome sequence prepared with more recent (so-called second-generation) sequencing technologies using a single yeast colony of Saccharomyces cerevisiae strain S288C has been published by Engel et al. (2013). The genome sequence of Saccharomyces cerevisiae enabled systematic genome-wide experimental approaches. The approach has been called a genome-scale systems analysis (Weissman et al., 2010). Systems analysis is a problem-solving technique that involves study of the entire set of parts or components that are connected to form the more complex whole process (the ‘system’).
Molecular analysis provides an overview of the complex network of gene regulatory events that are involved in a developmental (or any other) process, but it can be difficult to focus on the unique decisive events that characterise (and/or initiate) the process. New sequencing methods have hastened genome scale comparative studies of fungi by reducing the cost and difficulty of sequencing. This has enabled wider use of sequencing applications, such as to study genome-wide variation in populations (for example, Wyss et al., 2016) or to profile RNA transcripts totally (Conesa et al., 2016). The rapid impact of this level of analysis on research with yeast prompted efforts to expand genomic resources for other fungi, and the Fungal Genome Initiative was launched as an organised genome sequencing effort to assemble genomes of over 100 fungi in public databases to promote comparative and evolutionary studies across the fungal kingdom (Cuomo & Birren, 2010; Stajich, 2017). If you’ve not already done so, check out volumes 470, 500, 531 and 546 in the Methods in Enzymology book series; the citations are given above.
Following on from the development of new sequencing technologies, the pace of progress accelerated considerably, and in 2011 [http://mycor.nancy.inra.fr/blogGenomes/?p=2659], the 1000 Fungal Genomes Project [http://1000.fungalgenomes.org/home/] aimed to ‘…sequence 1000+ fungal genomes…’ and at the time of writing (2018), there are over 1000 fungal genomes listed as complete in at least draft form in the Mycocosm portal of the JGI (the U.S. Department of Energy (DOE) Joint Genome Institute; https://jgi.doe.gov/) [https://genome.jgi.doe.gov/fungi/] (Markowitz et al., 2012, 2015; Grigoriev et al., 2014; Nordberg et al., 2014) [and see JGI annual Progress Reports at https://jgi.doe.gov/about-us/progress-report/]. We will delve further into these general topic areas in Chapter 18, here, as an illustration of some of the extensive genomic systems analysis work that has been done, we will return to the Ink cap mushroom Coprinopsis cinerea, and particularly its fruit body development.
Coprinopsis cinerea was included in the list of fungi chosen for the Fungal Genome Initiative in 2002 (Cuomo & Birren, 2010), and the genome of Coprinopsis cinerea, which comprises 36 megabase pairs (a megabase pair (Mbp) is a unit of length of nucleic acids, equal to one million base pairs) was sequenced and assembled into 13 chromosomes by an international team of 47 researchers (Stajich et al., 2010). This was used to investigate how rates of meiotic recombination varied along the chromosomes, and the distribution, expression, and conservation of single-copy genes and gene families. Recently, a chromosome-level assembly has been completed of the homokaryotic Coprinopsis cinerea strain A43mut B43mut pab1-1 #326, which is a widely used experimental model for developmental studies in this mushroom-forming fungus (Xie et al., 2020). This assembly yielded 13 chromosomes and a mitochondrial genome, and has 15,250 annotated genes with a high synteny with the C. cinerea strain Okayama-7 #130 which was the first genome assembly.
It was evident that regions of the genome of Coprinopsis cinerea that have been conserved over evolutionary time show low rates of meiotic recombination and lack sequences such as tandemly repeated members of large gene families, and transposable elements that could promote recombination between non-homologous chromosomes (called ectopic recombination), which can cause chromosomal rearrangements. Single-copy genes with identifiable orthologous genes in other basidiomycetes (orthologous means the same function in different species and descended from a common ancestor) were also mostly located in low-recombination regions of their chromosome. On the other hand, paralogous genes (meaning genes related by duplication that may evolve new functions) were found in the highly recombining regions and near chromosome ends. These latter included families of hydrophobin genes (Section 6.8) and cytochrome P450 gene families that code for the terminal oxidases of the electron transfer chains.
The genome project also revealed two genes of the DNA (cytosine-5)-methyltransferase-1 family that catalyses transfer of methyl groups to specific CpG structures in DNA to form 5-methylcytosine, a process called DNA methylation. This confirms prior studies which showed methylation of repeated genes in Coprinopsis cinerea. DNA methylation is used to control gene expression by changing the activity of DNA without changing the sequence; methylation of a gene’s promoter typically represses transcription. The Coprinopsis cinerea genome contained 38 copies of a gene predicted to catalyse the formation of the additional modified base, 5-hydroxymethylcytosine, which has been detected in C. cinerea DNA. This is especially relevant to our current discussion of fungal development because 5-hydroxymethylcytosine has a role in the ‘poised (or bivalent) chromatin’ domains found in mammalian embryonic stem cells that play an important role in cell differentiation.
Poised (bivalent) chromatin is defined by the simultaneous presence of histone modifications associated with both gene activation and gene repression and is usually found near promoters of transcriptionally silent developmental regulatory genes. In general, pluripotent cells (capable of giving rise to several different cell types) have high numbers of poised domains compared with more differentiated cells, and these domains tend to resolve toward a purely active or purely repressed state during differentiation (Voigt et al., 2013; Lesch & Page, 2014).
5-Methylcytosine in DNA of eukaryotes is an important mark for epigenetic gene regulation; epigenetic marks silence certain gene sequences and activate others so that cells can differentiate. The ‘ten eleven translocation’ enzymes (TET; the enzymes are named for a common translocation in human cancers) oxidise 5-methylcytosines and promote locus-specific reversal of DNA methylation. ‘J binding proteins’ (JBP; originally named for their effect on glycosylated thymine in trypanosome DNA, which is known as base J) catalyse hydroxylation of bases in nucleic acids. TET/JBP enzymes are a family of nucleic acid base-modifying 2-oxoglutarate and iron-dependent dioxygenases that demethylate by converting 5-methylcytosine to oxidised methylcytosines, such as hydroxy-, formyl-, and carboxymethylcytosine, which serve as further epigenetic marks or intermediates for demethylation. Iyer et al. (2014) showed that fungi, especially all major clades of basidiomycetes, have numerous TET/JBP genes, which are often associated with a unique class of transposable elements. This pattern differs starkly from the situation in most other organisms that possess just a single or a few copies of the TET/JBP family. This unique transposon-TET/JBP association in fungi is likely to play important roles in speciation during evolution and epigenetic regulation. 5-methylcytosine (5mC) and oxidised methylcytosines (oxi-mCs) have been mapped in Coprinopsis cinerea at close to base-pair resolution (Chavez et al., 2014). Centromeres and transposable elements had distinctive patterns of 5mC and oxi-mC, and 5mC and oxi-mC within genes silenced duplicated genes.
The Coprinopsis cinerea genome includes several highly-expanded unusual protein kinase families including some orthologous to kinases of Saccharomyces cerevisiae, several kinase classes found in animals and/or Dictyostelium, but lost from yeast, and a hugely expanded, to 133 members, family of FunK1 kinases. Protein kinases are enzymes that regulate the activity of other enzymes by phosphorylation, which may turn an inactive enzyme into an active form (or vice versa), or otherwise change enzyme activity, cellular location, or its interaction with other proteins. FunK1 kinases are only found in multicellular ascomycete and basidiomycete fungi. The unusually large gene family in Coprinopsis cinerea shows considerable coding diversity; also 59 of the 133 FunK1 family genes are found within a short region near the end of chromosome 9, overlapping a recombination hot spot in a region where meiotic synapsis initiates. The FunK1 kinases are differentially expressed during dikaryon formation (mating), so they appear to have different functions during sexual morphogenesis and important roles development of Coprinopsis cinerea.
Finally, when the genomes of Coprinopsis cinerea and Laccaria bicolor, an ectomycorrhizal basidiomycete that grows in mixed birch and pine woods, were compared they were found to share extensive regions of synteny (meaning blocks of genes in a similar order within two sets of chromosomes that are being compared with each other). The largest syntenic blocks were found to occur in regions with low meiotic recombination rates, no transposable elements, and tight gene spacing (Stajich et al., 2010).
More evidence of tissue and development-specific gene control was found by studies that focussed on the transcriptional programme of Coprinopsis cinerea. Burns et al. (2010) examined the transcripts produced during meiosis, including time points extending over the 15 hours from before haploid nuclear fusion through to formation of basidiospore tetrads (see Section 8.4, Fig. 8.5 and Section 12.9). They found that about 20% of the genes in the genome are differentially regulated during this developmental process, with successive waves of transcription apparent in nine transcriptional clusters, one of which was enriched for meiotic functions. Transcriptional waves of this sort in other organisms have been found to represent differential expression of genes essential for recombination, chromosome cohesion, chromosome segregation, etc. Comparison with meiotic expression data from Saccharomyces cerevisiae and Schizosaccharomyces pombe showed that meiotic regulatory machinery was not conserved (not too surprising as, physiologically, both yeasts are stimulated to enter meiosis by nutritional limitation, while induction of fruit body development and meiosis in Coprinopsis cinerea is largely controlled by illumination and temperature).
However, a ‘core meiotic transcriptome’ of 75 genes was conserved between the three fungi. Ascomycetes and basidiomycetes are thought to have diverged in evolution about 500 to 900 million years ago, with Saccharomyces cerevisiae and Schizosaccharomyces pombe diverging from one another shortly after. Conservation of the core meiotic transcriptome between these three fungi implies that the essential transcriptional programme of meiosis has persisted through at least half a billion years of evolution, and potentially for up to a billion years (Burns et al., 2010).
The transcription pattern during the transition from the vegetative mycelium to the primordium during fruiting body development of Coprinopsis cinerea was examined by Cheng et al. (2013). They evaluated the expression of more than 3,000 genes in both growth stages and discovered that almost one-third of these genes were preferentially expressed in one or other stage. The data indicated that various structural and functional protein families were uniquely employed in one or other stage and that during primordial growth, cellular metabolism is highly up-regulated. Various signalling pathways were also up-regulated, particularly:
- the 'protein kinase A' family of enzymes whose activity is dependent on cellular levels of cyclic AMP (cAMP-PKA),
- the 'mitogen-activated protein kinase' family (MAPK)
- the ‘target of rapamycin’ (TOR) signalling pathway, which integrates both intracellular and extracellular signals and serves as a central regulator of cell metabolism.
All of which is consistent with the idea that sensing of nutrient levels and environmental conditions are important in this developmental transition. Interestingly, over 100 of the up-regulated genes were found to be unique to mushroom forming basidiomycetes, highlighting the novelty of fruit body development in these fungi (Cheng et al., 2013).
A particularly interesting example in the winter mushroom (Flammulina velutipes), is a transcription factor (LFC1) with negative regulatory function in basidioma development. Neither overexpression nor knockdown of lfc1 affected hyphal vegetative growth, but knockdown of lfc1 could promote basidioma development and shorten cultivation time by 2 days, while overexpression of lfc1 delayed the optimal harvest time by 3 days. RNA-seq analysis revealed that 455 genes had opposite transcriptional responses to lfc1 overexpression and knockdown: these included genes encoding hydrophobins, lectins, cytochrome P450 and membrane components. As LFC1 homologues are widely present in mushroom species, this is thought to represent a general basidioma development pathway (Wu et al., 2020).
The transcriptomes of stage 1 primordia and vegetative mycelium of Coprinopsis cinerea were also studied by Plaza et al. (2014), but these authors paid attention to regulation of the expression of defence genes as well as the conserved transcriptional patterns of sexual reproduction. Comparing their results with analogous data from Laccaria bicolor and Schizophyllum commune revealed a conserved transcriptional circuitry in basidiomycete fruit bodies consisting of nearly 70 genes involved in mushroom development and function. They also found evidence for a role of the ‘Velvet protein regulon’ in sexual development of basidiomycetes, as a velvet domain-containing protein was found to be up-regulated in all three fungi. The four velvet proteins VeA, VelB, VelC, and VosA were originally identified in Aspergillus nidulans and shown to form a family of fungal regulators that couple together light-regulated processes, control of secondary metabolism, growth and differentiation. Velvet proteins share a region of about 150 amino acids, the velvet domain, which lacks sequence homology to any other proteins (Bayram et al., 2008; Ahmed et al., 2014). These velvet regulators are present in most clades of the fungal kingdom from chytrids to basidiomycetes. Velvet proteins have presumably been recruited into the basidiome transcriptional circuit as they are one of the most ancient systems regulating the production of asexual spores in fungi (Etxebeste et al., 2019). In contrast, the central regulator BrlA (discussed in Section 9.4 about Aspergillus conidiophores) emerged later in fungal evolution, with the class Eurotiomycetes (see Section 3.7).
Muraguchi et al. (2015) extended the investigation of the transcriptome during fruit body development in Coprinopsis cinerea by using high-throughput sequencing of cDNA libraries from 13 developmental points (nine developmental stages, but from stage 6 (onset of meiosis) cap and stipe tissues were analysed separately; see Table 12.1). The analysis revealed changes in expression of many differentially expressed genes between two succeeding developmental stages and between vegetative mycelium and each developmental stage. GO analysis of differentially expressed genes (DEGs) in each transition revealed an overall developmental framework and many notable cellular events; these results are summarised in Fig. 28 and Table 1 [CLICK HERE to download a PDF of these two items].
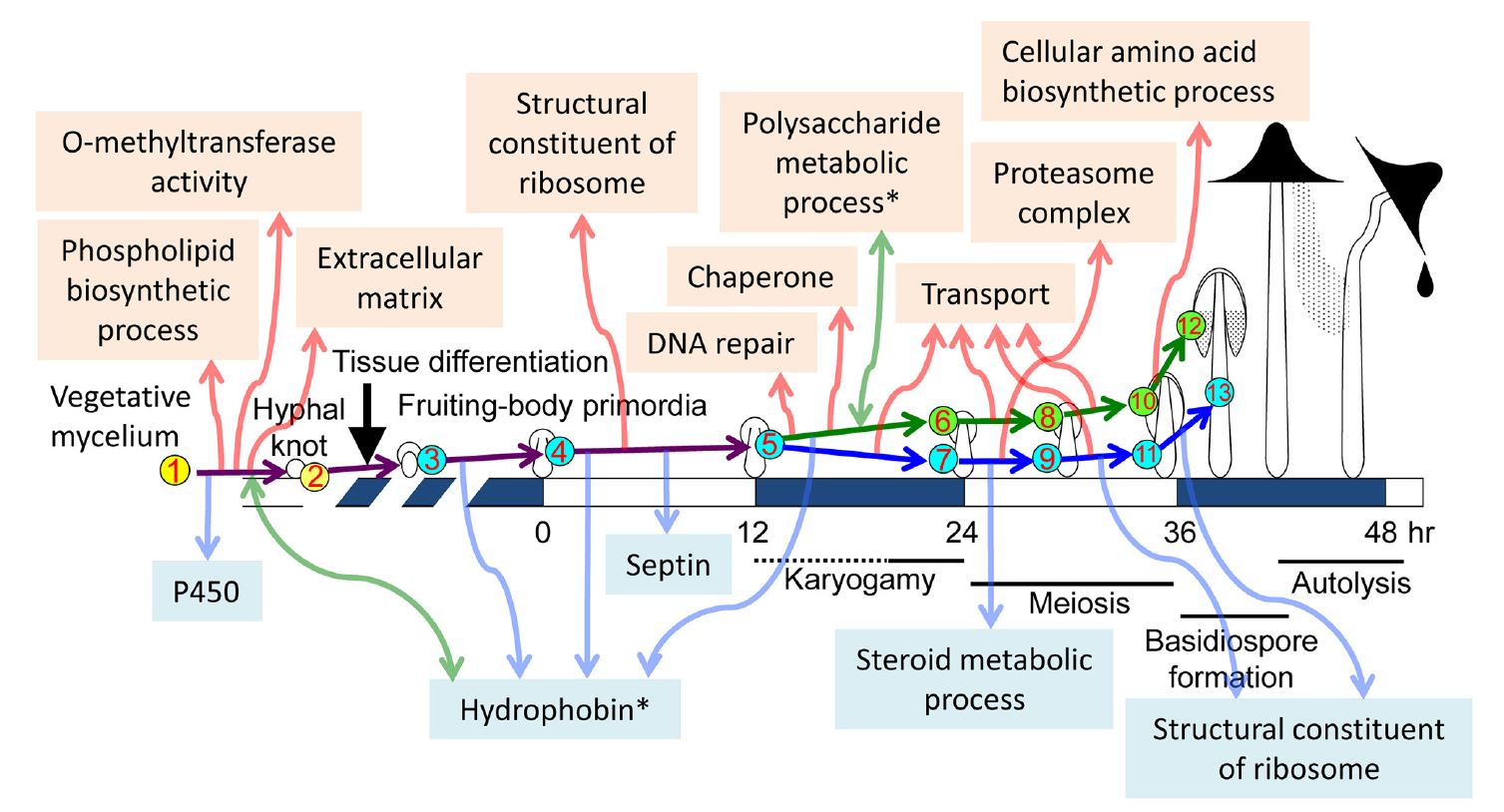 |
| Fig. 28. Summary of major
transcriptional events during morphogenetic transitions during fruit body
development in
Coprinopsis cinerea. The cartoon diagrams of fruiting across the
centre run from vegetative mycelium at left to
autolysing mature fruit body at right, and are
placed on an average time scale with time zero being set by the illumination
cycle that triggers primordium maturation. The nine developmental stages selected to investigate the transcriptome are shown with numbers in circles (cap and stipe tissue samples were taken separately from 24 h onwards). Developmental and cellular events in each stage and tissue are shown in Table 12.1. The study identified differentially expressed gene sets using GO enrichment analysis [http://geneontology.org/page/go-enrichment-analysis]; that is, sets of gene sequences of related function that are up-regulated or down-regulated in pairwise comparisons between two succeeding developmental stages, and between vegetative mycelium and each individual developmental stage. The program does this by statistically determining which sequence(s) are over-represented (or under-represented) in each member of the pair relative to the other. Notable transcriptional programme events are depicted in the text labels. Events detected by up-regulated differentially expressed genes are indicated in the upper part with upward red arrows; down-regulated differentially expressed gene sets are shown in the lower part with downward blue arrows. Sets which featured a mix of both up-regulated and downregulated gene sequences of related function are indicated with asterisks and bidirectional green arrows. Taken from Muraguchi et al. (2015) under Creative Commons licence https://creativecommons.org/licenses/by/4.0/. |
Table 1. Major morphogenetic events and transcriptional transitions during fruit body development in Coprinopsis cinerea
2, sets which featured a mix of both up-regulated and downregulated differentially expressed gene sequences of related function are indicated with this bidirectional arrow and green text. *, indicates that only the first 3,000 genes were analysed. Data from Muraguchi et al. (2015) [https://doi.org/10.1371/journal.pone.0141586]. CLICK HERE to download a PDF version of this Table (with its associated figure). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Looking at the discussion so far you can see that the conclusions from the pregenomics era have been broadly confirmed by genome systems analysis, though expanded and given their quantitative context (Table 1). Several hundred to a thousand genes are preferentially expressed at different stages in development of the fruit body. Various signalling pathways have been implicated including cAMP-PKA, MAPK and TOR, the fungus-specific velvet protein regulon and the FunK1 kinases, which are only found in multicellular ascomycete and basidiomycete fungi. Considering the accounts given above (Section 12.11) of the cycles of glycogen accumulation and translocation in different fruit body tissues it is interesting to see in Fig. 28 that the category ‘polysaccharide metabolic process’ is a set that featured a mix of both up-regulated and down-regulated transcripts. Indeed, transcriptional profiling is beginning to unravel this ‘glycogen regulatory circuit’ by showing that glycogen synthase kinase 3 (GSK-3) activity is essential for fruit body formation in Coprinopsis cinerea by directly affecting expression of fruit body induction genes together with genes in downstream cellular processes (Chan et al., 2018); and experimental inhibition of GSK-3 expression can control fruit body development (Chang et al., 2018b).
Similarly, the importance attached to enzymes involved in ammonium scavenging and the urea cycle (Section 12.11) fits well with the up-regulation of transcripts in the category ‘cellular amino acid biosynthetic process’. There are several other features of Fig. 28 that confirm deductions from the pre-genomics era, particularly the up-regulation of ‘transport’ transcripts in both cap and stipe between stages 5 and 6/7, and then again between 6/7 and 8/9, and yet again between 8/9 and 10/11 all confirm the presumed rapid translocation of metabolites and water during fruit body maturation. Perhaps not quite so obvious is that the up-regulation of transcripts in the ‘extracellular matrix’ category fits well with what was known about the importance of the extracellular matrix to hyphal knot formation; and the up-regulation of ‘proteasome complex’ in the stipe between stages 6/7 and 8/9, with the implication that protein degradation is important to the stipe at these stages for some reason is compatible with what is known about the biochemistry of the Coprinopsis stipe which, unusually, does not accumulate sugars or polyols and is likely, therefore to use the carbon skeletons of amino acids derived from protein degradation as energy source. Furthermore, the conclusion that differentiation of fungal multicellular structures is genetically compartmentalised with normal morphogenesis being made up of several-to-many developmental subroutines (see above in this Section) are equivalent to the transcriptional clusters, transcriptional waves of differential gene expression and differentially expressed gene sets detected in these different studies of the transcriptional programme of the Coprinopsis cinerea genome.
Differential transcriptional patterns of a family of glycoside hydrolases of Coprinopsis cinerea matched well with the requirement for the wall-softening that these enzymes effect in the germination of basidiospores, hyphal growth and branching, primordium formation, stipe elongation, pileus expansion and autolysis (Kang et al., 2019b). Some members of the family were predominantly expressed in dormant basidiospores, primordia and during basidiosporogenesis. Other members were dominantly expressed in the germinating basidiospores, growing mycelia, and elongating stipes. Stipe elongation results from elongation of existing stipe cells. Chitinases play a key role in this stipe wall remodelling, whilst β-glucanases cooperate with chitinase to induce stipe wall extension. In addition, fungal expansin-like proteins can bind to chitin to enhance chitin hydrolysis (expansins are small proteins in plants that loosen plant cell walls in a non-enzymatic but pH dependent manner, reducing adhesion between adjacent wall polysaccharides and allowing local sliding of wall polymers) (Liu et al., 2021). The dynamics of the proteome during fruit body development in C. cinerea has also been studied (Majcherczyk et al., 2019).
Muraguchi et al. (2015) also observed dramatic changes in gene expression of some transcription factor networks, and several examples of natural antisense transcripts (NATs) that are likely to be involved in developmental regulation of fruiting. NATs are widespread in eukaryotes; they are RNAs that are complementary to other (‘sense’) RNA transcripts. Evidence indicates that sense and antisense transcripts interact, suggesting a role for NATs in the regulation of gene expression (Wight & Werner, 2013). MicroRNAs (miRNAs) are about 22 nucleotides in length and have essential roles in post-transcriptional regulation of gene expression in animals and plants. Although little studied in fungi, microRNAs have been associated with early fruit body development in Coprinopsis cinerea, where Lau et al. (2018) established their potential targets and demonstrated differential miRNA expression during the early developmental stages of the mushroom, suggesting their involvement in regulating development in this fungus.
Krizsán et al. (2019a & b) constructed a reference atlas of mushroom formation based on comparisons of developmental transcriptome data of more than 200 whole genomes belonging to six species of Agaricomycetes (Armillaria ostoyae, Coprinopsis cinerea, Lentinus tigrinus, Phanerochaete chrysosporium, Rickenella mellea and Schizophyllum commune). 12,003 to 17,822 expressed genes were detected in the six species of which 938 to 7,605 were developmentally regulated. Importantly, nearly 300 conserved gene families in more than 70 functional groups contained developmentally regulated genes. These data outline the major multicellularity-related and developmental processes of mushrooms, including the role of transcriptional reprogramming, gene coexpression networks (called ‘modules’ by the authors but corresponding to the developmental subroutines discussed above), and alternative splicing. Many of the developmentally regulated sequences were expanded in multicellular plants and/or animals too, implying convergent evolution towards solutions to the challenges imposed by complex multicellularity within independently evolving lineages. Data relating to the evolutionary age distribution of developmentally regulated genes suggested that most developmental gene families are older than fruit body formation; the sequences falling into three groups (a) genes shared by all living species, (b) genes shared by the Dikarya (Ascomycota and Basidiomycota), and (c) species-specific genes.
Another promising technique for detecting transcriptional regulatory relationships on a genome-wide scale that is beginning to be applied to fungal development is the analysis of expression Quantitative Trait Loci (eQTL). A quantitative trait locus (QTL) is a DNA segment associated with a particular phenotype, which varies across a population in a manner attributed to polygenic control (presumed to be the product of two or more genes together with their environment). The approach is essentially a metagenomic statistical analysis of variation in the phenotype of interest that can reveal the genetic architecture of that trait; that is, whether phenotype is controlled by many genes of small effect, or by a few genes of large effect. Expression quantitative trait loci (eQTLs) are genomic loci that describe the variation in expression levels of mRNA molecules. Analysis of eQTLs can recognise potential regulatory factors of gene expression and could identify master regulators in the genome (Michaelson et al., 2009; Liseron-Monfils & Ware, 2015; Cheng et al., 2016; Chang et al., 2018a).
Because the current interpretation of the mechanism of gravity perception in the Coprinopsis cinerea fruit body is that cytoskeletal interactions communicated to the transmembrane domains of a sugar transporter change its substrate specificity to cause a 6-deoxy hexose sugar growth regulator to be transported asymmetrically (see Section 9.8 and, especially, the discussion of the possible mechanism of Fungiflex activity in Section 12.9), it is significant that the eQTL network analysis of Chang et al. (2018a) identified a sugar transporter as an ‘eQTL hotspot’. Such hotspots are believed to regulate expression of several genes, and the sugar transporter identified as a hotspot in this study regulated the expression of at least 12 associated genes. One of those associated genes specifies a Zn-finger protein (which can bind DNA, RNA, proteins or small molecules) and another is a pyranose dehydrogenase enzyme with broad substrate tolerance for oxidation of different sugars. The transporter is thought to interact with the pyranose dehydrogenase via a transcription factor.
We already knew about the extensive use of transcription factors to direct transcription to specific genes in fungi (see above, Sections 2.4; 5.4; 5.13; 6.7; 8.5; 9.4), particularly the homeodomain proteins of the mating type factors (Bobola & Merabet, 2017; Vonk & Ohm, 2018), so add those to the list of fungal regulators. We should also add to that list of regulators translational triggering and feedback fixation, discovered in the 1990s in research on Aspergillus nidulans conidiophores (Section 9.4), which solve the problems of how to respond to a transient signal, and how to maintain that response when the signal has dissipated. The mechanism involves a genetic sequence, in this case the brlA locus, that codes for an RNA transcript containing overlapping reading frames (Fig. 9.9). Alternative splicing of precursor mRNA is now established as an essential mechanism to increase the complexity of gene expression that plays an important role in cellular differentiation and development in eukaryotes (Wang et al., 2015). Alternative splicing is common in higher plants and animals and increases the coding capacity from a limited set of genes. Grützmann et al. (2014) claim that alternative splicing is also a common phenomenon in fungi, and that, on average, about 6.0% of genes in ascomycetes, and 8.6% of basidiomycete genes show alternative splicing (Cryptococcus neoformans had the extraordinary rate of 18%), and that this process is associated with multicellular complexity, dimorphic switching, stress response and pathogenic virulence.
Fungal mycelia, fruit body primordia and mature fruit bodies are attractive sources of food to small animals including molluscs, arthropods and nematodes (Section 11.2; Fig. 11.11), and it is well known that mushrooms produce defence proteins and secondary metabolites to deter such predators and other competitors. Plaza et al. (2014) revealed the existence of two different sets of fungal defence proteins in the proteomes of fruit body primordia of Coprinopsis cinerea. One of the primordium-specific proteins is toxic to insects and nematodes and the expression pattern of these proteins correlates with the type of antagonists with which these tissues are confronted. For instance, cytoplasmic lectins, also referred to as fruit body lectins due to their specific expression pattern, have been shown to be toxic to nematodes, insect larvae and amoebae (Bleuler-Martínez et al., 2011; see Section 15.6). In contrast, vegetative mycelia secrete antibacterial peptides which deter competing bacteria. Known as defensins, these are found in vertebrates, higher plants and invertebrates as well as saprotrophic fungi (Wu et al., 2014). Defensins have therapeutic promise; plectasin, isolated from the ascomycete Pseudoplectania nigrella, is especially active against Streptococcus pneumoniae, including strains resistant to conventional antibiotics. Genome editing techniques are now available to manipulate production of potentially useful metabolites like these (Doudna & Sontheimer, 2014; and see Section 18.14), and a novel method of high-throughput CRISPR/Cas9-based genome editing of Coprinopsis cinerea using cryopreserved protoplasts has been reported (Sugano et al., 2017).
Surprisingly, there is no mention in any of these studies of homologues of sequences of the animal signalling mechanisms known as Wnt, Hedgehog, Notch and TGF-α, all of which are essential and highly conserved components of normal development in animals. Systematic mining of fungal genomes for these sequences established that all were absent from the fungal genomes, just as they are absent from plants (Moore et al., 2005). Subsequently, a fully comprehensive data-mining exercise searched for homologies to all developmental sequences in all sequenced genomes, animal, fungal and plant that were then available (Moore & Meškauskas, 2006; 2009). The outcome was that homology between the genomes of fungi and other eukaryotes was limited to 78 sequences involved in the architecture of the eukaryotic cell. This survey demonstrated that there was no evidence in fungal genomes of Wnt, Hedgehog, Notch, TGF, p53, which are all crucial animal regulators, nor of the plant control sequences SINA, or NAM. All of which leads to the conclusion that the unique cell biology of filamentous fungi has caused them to evolve control of their multicellular development in a radically different fashion from that in animals and plants.
In this Section we have focussed on the developmental biology of Coprinopsis cinerea but as we indicate above in this Section ‘…there are over 1000 fungal genomes listed as complete in at least draft form in the Mycocosm portal…’ We can’t prolong this discussion any further, so to ease you into the world of fungal genomics we’ve put together a list of publications (open access text favoured, so you should be able to obtain the full texts) in the following Resources Box.
Resources Box Genomic systems analysis of fungi MycoCosm is a fungal genomics portal (http://jgi.doe.gov/fungi), developed by the US Department of Energy Joint Genome Institute to support integration, analysis and dissemination of fungal genome sequences and other ‘omics’ data by providing interactive web-based tools. MycoCosm also promotes and facilitates user community participation through the nomination of new species of fungi for sequencing, and the annotation and analysis of resulting data. View the current list on the following page, and links to publications about the sequences: https://genome.jgi.doe.gov/fungi/fungi.info.html. Grigoriev, I.V., Nikitin, R., Haridas, S., Kuo, A., Ohm, R., Otillar, R., Riley, R., Salamov, A., Zhao, X., Korzeniewski, F., Smirnova, T., Nordberg, H., Dubchak, I. & Shabalov, I. (2014). MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Research, 42: D699-D704. DOI: https://doi.org/10.1093/nar/gkt1183. |
|
We have prepared a list of published papers
(additional to those cited in the text) that will ease you into the
world of fungal genomics. The list has been put together favouring these
criteria: (a) open access publishing; and (b) illustrating different
approaches.
CLICK HERE to download the list as a PDF And check out this special issue of the journal Fungal Genetics and Biology (Volume 89, pages 1-156, April 2016) entitled The Era of Synthetic Biology in Yeast and Filamentous Fungi at https://www.sciencedirect.com/journal/fungal-genetics-and-biology/vol/89/suppl/C. The Earth BioGenome Project aims to sequence, catalogue, and characterise the genomes of all of Earth’s eukaryotic biodiversity, as described in the following: Lewin, H.A., Robinson, G.E., Kress, W.J., Baker, W.J., Coddington, J., Crandall, K.A., Durbin, R., Edwards, S.V., Forest, F., Gilbert, M.T.P., Goldstein, M.M., Grigoriev, I.V., Hackett, K.J., Haussler, D., Jarvis, E.D., Johnson, W.E., Patrinos, A., Richards, S., Castilla-Rubio, J.C., van Sluys, M.-A., Soltis, P.S., Xu, X., Yang, H. & Zhang, G. (2018). Earth BioGenome Project: Sequencing life for the future of life. Proceedings of the National Academy of Sciences of the United States of America, 115: 4325-4333. DOI: https://doi.org/10.1073/pnas.1720115115. See also the Earth BioGenome Project website at https://www.earthbiogenome.org/ and the Darwin Tree of Life Project at https://www.sanger.ac.uk/news/view/genetic-code-66000-uk-species-be-sequenced. |
Updated May, 2021