4.1 Origin of drugs in current use: the cyclosporine story (contributed by Harriet Upton, 2001)
Abstract
Discovery of cyclosporine in 1971 began a new era in immunopharmacology. It was the first immunosuppressive drug that allowed selective immunoregulation of T cells without excessive toxicity. Cyclosporine was isolated from the fungus Tolypocladium inflatum. Cyclosporine was first investigated as an anti-fungal antibiotic but its spectrum was too narrow to be of any clinical use. J. F. Borel discovered its immunosuppressive activity in 1976. This led to further investigations into its properties involving further immunological tests and investigations into its structure and synthesis. Cyclosporine has unwanted side effects, notably nephrotoxicity. Animal testing showed cyclosporin to be sufficiently non-toxic to begin clinical trials. These initially failed due to poor absorption of the drug. Once this had been overcome, results were encouraging enough for cyclosporine to be licensed for use in clinical practice. There is some controversy between Borel and other workers over priority in the discovery of cyclosporine and its pre-clinical development, which is examined in this review. Cyclosporine changed the face of transplantation. It decreased morbidity and enabled the routine transplantation of organs that until then had only been done experimentally.
Introduction
We all know the story about the discovery of Penicillin, but what about other drugs now in common use? This article investigates the events that led to the discovery of cyclosporine [initially known as cyclosporin A, now known as ciclosporine in Europe and cyclosporine in the USA], and the developments that resulted from its introduction into clinical practice.
Naming cyclosporine
Cyclosporine is used clinically to suppress the immune response in transplant patients and prevent organ rejection. You are likely to find this drug given any of three nonproprietary names:
cyclosporine was assigned by the United States Adopted Names Council, which is co-sponsored by the American Medical Association (AMA), the United States Pharmacopeial Convention (USP), and the American Pharmacists Association (APhA).
Cyclosporin (without the terminal ‘e’) is a British Approved Name (BAN) as defined in the British Pharmacopoeia (BP) and is used in many countries around the world, especially those of the Commonwealth of Nations.
European Union legislation requiring harmonisation of the BP with the European Pharmacopoeia (EP) has resulted in use of the name ciclosporin in the UK. This is an International Nonproprietary Name (INN) designated by the World Health Organisation (WHO) and with the exception of adrenaline/epinephrine, BANs are now the same as the INNs.
Of course, there are even more proprietary names!
Irene
A man who put a soil sample in a plastic bag saved the life of Irene, a vivacious student, who, at the age of 18 years was diagnosed with acute myeloblastic leukaemia. It progressed rapidly and severely. Chemotherapy might offer her a brief remission but would inevitably end in a fatal relapse. However Irene had two pieces of good fortune. Firstly, she was immunonologically (HLA) compatible with her younger sister. Secondly cyclosporine had been discovered a few years previously. Doctors expressed concern about cyclosporine toxicity and about likely development of the devastating host-versus-graft disease. Despite their anxieties there was no other hope for Irene and a bone marrow transplant was undertaken. Irene developed severe renal complications. Little was known about cyclosporine nephrotoxicity and she was probably given excessively high doses. But thanks to cyclosporine she only suffered from a mild host-versus-graft reaction and was able to leave hospital to live a normal life [1].
What is cyclosporine and what are its uses?
Today organ and bone marrow transplants are routinely performed. Cyclosporine is still used to treat the rejection reactions that occur when a foreign organ is attacked by the body’s immune system. Cyclosporine is a fungal peptide, isolated from Tolypocladium inflatum Gams. It was the first immunosuppressant that acted selectively to suppress T-cell immunity.
Cyclosporine is at present (March 2001) approved for use in organ transplantation to prevent graft rejection in kidney, liver, heart, lung and combined heart-lung transplants. It is used to prevent rejection following bone marrow transplantation and in the prophylaxis of host-versus-graft disease. It is also used in the treatment of psoriasis, atopic dermatitis, rheumatoid arthritis and nephrotic syndrome.
Why was the discovery of cyclosporine so important?
Discovery of immunosuppression by cyclosporine in 1976 is attributed to J. F. Borel (Fig. 5). In 1983 cyclosporine was approved for clinical use to prevent graft rejection in transplantation. Most of the surgical problems of allograft transplantation had already been solved by this time.
Since 1961 the standard method of achieving immunosuppression had been a combination of azathioprine and corticosteroids. Azathioprine inhibits cell proliferation non-selectively. Its main unwanted side effect is depression of the bone marrow, other toxic effects include increased susceptibility to infections, a mild hepatotoxicity, skin eruptions, nausea and vomiting. Corticosteroids inhibit T lymphocytes and have an anti-inflammatory effect. Side effects include diabetes, avascular necrosis of bones and increased tendency to infections [2].
Cyclosporine was the strongest immunosuppressor to be discovered at the time; it also overcame many of the risk factors associated with azathioprine and is relatively non-toxic to bone marrow. With the introduction of cyclosporine patient morbidity fell. It became possible to transplant organs with a one year success rate 20% higher than previously [3], and to transplant organs successfully which previously had only been done in experimentation: the heart, the liver, the lung and combined heart lung transplants [4].
As well as transplantation, cyclosporine has been used in most autoimmune diseases. In the 1980’s experimental treatment with cyclosporine of insulin-dependent diabetes mellitus, inflammatory bowel disease, chronic asthma, atopic dermatitis, aplastic anaemia and psoriasis supported evidence of their T cell mediated nature [5].
The discovery of cyclosporine
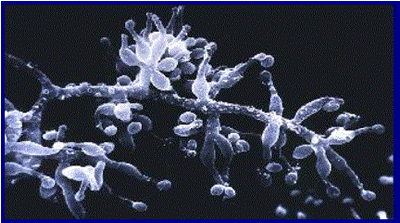
Discovery of an anti-fungal antibiotic. A tradition established as part of a programme set up in 1957 to search for new antibiotic drugs from fungal metabolites was for Sandoz employees on business trips and holidays to take plastic bags with them for collecting soil samples that were catalogued and later screened. In March 1970 in the Microbiology Department at Sandoz Ltd. (Basel), a Swiss pharmaceutical company, the fungus Tolypocladium inflatum Gams (Fig. 1) was isolated by B. Thiele from two soil samples, the first from Wisconsin, USA and the second from the Hardanger Vidda in Norway. These soil samples had been collected by Sandoz employees.
Fig. 1. Scanning electron micrograph of Tolypocladium inflatum
The Microbiology Department at Sandoz had developed a computer-aided evaluation programme for screening and evaluating sampled fungi [6].The programme enabled rapid evaluation of the samples, recognising and eliminating common fungi, and related strains, that produced known compounds from further study. This meant that more time could be spent evaluating the rare fungi in the samples, which would be more likely to produce metabolites that had potential new antibiotic activity. The programme identified Tolypocladium inflatum; it was previously unknown to the Sandoz team and produced interesting metabolites.
Next, Z.L. Kis routinely isolated a metabolite mixture from Tolypocladium inflatum. The characteristics were fed into another computer program based on an exhaustive file of data extracted from the literature [6] and showed the presence of a group of metabolites, which were new to Sandoz. These metabolites were found to possess sufficient antifungal activity to warrant further attention.
For further investigation larger samples were required. Tolypocladium inflatum was cultivated in a fermenter and a technique for the isolation of two of the metabolites later named cyclosporine A and C was developed [7]. Samples were produced in submerged culture and extracted by organic solvents. M. Dreyfuss and colleagues examined the antibacterial and anti-fungal activity of cyclosporine A and C. They were found to have only a narrow spectrum of activity against fungi, and no antibacterial activity was found.
Only a few species of yeast were found to be sensitive to the metabolites. When grown in solid media and in contact with cyclosporine the growth rates of the sensitive species was decreased. Strains of some Mucorales, ascomycetes and fungi imperfecti showed sensitivity to variable degrees, the inhibition taking the form of deformation and branching of growing hyphal tips. There was no effect on germination of spores or conidia of the affected fungi.
By analysing and comparing the taxonomic positions of sensitive organisms, Dreyfuss and colleagues hypothesised that the mode of anti-fungal action was due to an inhibition of cell wall synthesis, in particular chitin synthesis. Cyclosporine activity was compared to the only known chitin blocking antibiotic Polyoxin, which exhibited a similarly narrow spectrum to that observed in cyclosporine [7].
An anti-fungal drug that inhibited cell wall synthesis would have been a useful discovery, as it would have high specificity and low toxicity to non-fungal hosts, similar to the indispensable group of β-lactam antibiotics. However, cyclosporine was found to be inactive against Sporobolomyces roseus and Sporobolomyces antarcticus ( Table 1).This indicated a different mode of action for the cyclosporines.
Table 1. Extent of inhibition zones in fungal growth (measured in mm) caused by some antifungal agents
Organism
Polyoxin A
cyclosporine A
C
Neurospora crassa
Neurospora crassa ‘slime’ mutant
0
Rhodotorula rubra
Sporobolomyces roseus
Sporobolomycesantarcticus
Due to their narrow spectrum and weak action there was no future for cyclosporine as an antifungal agent and Dreyfuss and his colleagues stopped their investigations.
Discovery of immunosuppressive activity by cyclosporine
The first non-steroidal immunosuppressive, non-toxic to bone marrow, was discovered at Sandoz in 1962 in the search for new and useful fungal metabolites. It was isolated in 1965 from Pseudeurotium ovalis and was named Ovalacin. It depressed the immune response strongly but did not affect division of intestinal epithelial cells or myeloblast proliferation, unlike other cytostatic drugs. Ovalacin preceded the discovery of cyclosporine and was crucial for setting the stage for the latter's discovery. Ovalacin is 600 times more potent than cyclosporine by weight, but it failed clinical trials because of its toxic effects.
In January 1970 the head of the Pharmacology Department at Sandoz, K. Saameli, developed a programme of about 50 pharmacological tests performed by different Groups in the Pharmacological Department: the ‘General Screening Programme’ [8]. A. Rüegger from the Chemistry Department, aware that microbial metabolites often possess interesting pharmacological activity, submitted cyclosporine for the General Screening Programme in 1971. The sample was given the preparation number 24-556, and it was later found to contain mainly cyclosporine.
Out of all the pharmacological tests in the General Screening Programme only one produced a positive result. This was a test for immunosuppression. On day one, mice were intravenously injected with sheep erythrocytes and preparation 24-556 was injected intraperitoneally on the next four days. On day 7 a sample of serum was taken and titrated for antibodies. The initial results showed a decrease in haemoglutination by a factor of 1024 in comparison with the controls [8]. In the same mice no non-specific antiproliferative activity was found. These results gave the first indication that cyclosporine might be an important compound. The only other effects found by the General Screening programme were a mild analgesic action and nephrotoxicity in rats given high doses of preparation 24-556 over one week.
The General Screening Programme had discovered the three properties of cyclosporine which have both shaped and limited its future use. First was its immunosuppressive activity, second it has no non-specific cytostatic action and finally its nephrotoxicity.
Research and development of cyclosporine
After the initial interesting and promising results of the General Screening programme, further microbiological, chemical and pharmacological work was carried out at Sandoz. The initial experiment that had highlighted the immunosuppressive activity of cyclosporine (see above) was repeated but the results were disappointing. Administration of preparation 24-556 orally and by the intraperitoneal route only showed a four-fold decrease in haemoglutination, despite the use of a higher dose. If this had been seen in the original experiment further development of cyclosporine would never have been carried out. The low results were later found to have been due to poor absorption of the drug. An alternative procedure had been used to solubilise the highly hydrophobic cyclosporine. The hydrophobic nature of the drug was to cause more problems in cyclosporine’s subsequent development.
Luckily, other immunological assays that were routinely used at Sandoz, that had been developed for investigation of Ovalacin, showed activity. Further experiments showed that cyclosporine selectively inhibited the proliferation of lymphocytes by acting on an unknown and unique step in the process, whilst not affecting proliferation of other somatic cells. In the words of Borel: ‘It was almost too beautiful to be true’ [6].
The scientists who had developed cyclosporine this far were already convinced of its relevance to immunosuppression. However, the goals at Sandoz had changed by 1973; immunology was no longer regarded as a fertile research field. This change was due to the rapid developments in immunology that had taken place elsewhere. Although basic knowledge of immunology had improved dramatically, comparable improvement in clinical applications for this knowledge had not been developed. Organ transplantation was a small, unattractive market restricted mainly to kidney transplants with the use of cheap immunosuppressive drugs (such as azathioprine and corticosteroids ). It was estimated that $250 million would have been needed to develop cyclosporine through to US Food and Drug Administration approval. The recent failure of Ovalacin in its clinical trials was another factor that had led management to believe prospects for a new immunosuppressant were low. A way of getting approval for further development was therefore found. The scientists’ exact method for gaining official approval of cyclosporine further development is unclear; cyclosporine anti- chronic inflammatory action seems to have been a key element, however.
Whether further development was granted for preventing the symptoms of experimental encephalomyelitis in rats, as claimed by Stähelin [8] or to test on adjuvant arthritis in the rat (Borel & Kis [6]) is unclear. However, official permission to carry on with cyclosporine was indeed granted. The next step was to determine the exact structure of the active metabolites from Tolypocladium inflatum that were present in preparation 24-556.
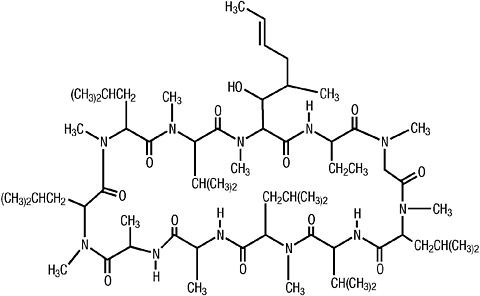
Fig. 2. The structure of cyclosporine.
The active metabolite was found to be a cyclic undecapeptide that was subsequently named cyclosporine. The structure and conformation of cyclosporine (Fig. 2) was determined by chemical degradation together with an X-ray crystallographic analysis of an iodo- derivative and by two-dimensional nuclear magnetic resonance (NMR) imaging studies of cyclosporine itself [9]. Cyclosporine was found to be rich in hydrophobic amino acids, neutral, insoluble in n-hexane and water but very soluble in all other organic solvents [10].
Chemical degradation. Cyclosporine was hydrolysed and was found to be made up of eleven amino acids, ten of which were known but the amino acid at position one was unknown [11].
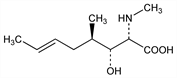
X-ray crystallographic analysis [10]. Cyclosporine was hard to cystallise on its own and so was initially analysed as crystalline iodocyclosporine. The unknown amino acid was found to have an R group structure as shown in Figure 3. It was found to be a beta-hydroxy, singly unsaturated amino acid (4R)-4[(E)-2-butenyl]-4,N-di-methyl-L-threonine, abbreviated to MeBmt.
Fig. 3. The R group of MeBmt
NMR spectra of cyclosporine [12]. NMR was used to determine the conformation of cyclosporine in a crystalline state and in solution. A later development, in 1984, namely the total synthesis of cyclosporine [13], enabled a systemic study of cyclosporine structure-activity relationships. Biological activity was found to be associated with amino acids 1, 2, 3, 4,10, and 11 which are on the surface of the molecule [14].
Two studies were performed to determine whether cyclosporine action was selective for lymphocytes and to exclude any cytostatic effects on cells other than lymphocytes. The first study showed that in vitro cyclosporine was 300 times more active in preventing the proliferation of spleen lymphoid cells than on nonlymphoid mastocytoma cells [15]. The second study examined the effect of cyclosporine on bone marrow cell count and haematopoietic myeloid stem cell proliferation in mice. Effects were minimal. Even at high doses, no effect was found on haematopoietic myeloid stem cell proliferation and bone marrow cell count was only slightly reduced. These studies proved without doubt the value of cyclosporine. The results were published in 1976 in Agents and Actions as ‘Biological effects of cyclosporine A: a new antilymphocyte agent’ [16]. It bought the discovery of cyclosporine to the world's attention.
The properties of cyclosporine interested Roy Y. Calne and his coworker D. G. White who had been involved in the development of azothioprine for transplantation. Calne (now Sir Roy; see Fig. 4 below) is well known as a pioneer of transplantation surgery. It was therefore in Cambridge that the first animal testing outside Sandoz was carried out.
Fig. 4. Sir Roy Calne
Fig. 5. Jean Francois Borel
Fig. 6. Hartman Stähelin
Toxicological studies. In 1975 toxicology studies were carried out at Sandoz in preparation for human testing. Rats given preparation 24-556 at high doses for 13 weeks showed renal and hepatic toxicity. In a similar experiment, dogs were treated with high doses of cyclosporine powder given orally in capsules, but showed no effects. Since the test on dogs was unrevealing, another toxicity study with cyclosporine was soon begun in the autumn of 1975, but this time in monkeys [8]. In these animals, the drug exhibited some activity, which led to the decision to begin clinical trials. The reason for the failure in the dog study was a low absorption of the cyclosporine powder. The results were sent to Calne, who in his animal experiments administered cyclosporine dissolved in olive oil. Calne’s results, especially in orthotopic heart grafts in the pig [17] were very encouraging (Table 2).
2. Survival times for orthotopic heart grafts in pigs with different immunosuppression treatments
Immunosuppression treatment
Mean survival
(days)
None
5.7
Azathioprine & methylprednisolone
14.7
Cyclosporine A
>67.8
range 22 to >111
Calne found that cyclosporine was a more effective immunosuppressive than any other drug that he had used in pigs with orthotopic cardiac allografts. In the discussion of his results he wrote, ‘sufficiently non-toxic and powerful as an immunosuppressant to make it [cyclosporine] an attractive candidate for clinical investigation in patients receiving organ grafts.’
Clinical trials. The first human trials were started in late 1976. Pure undissolved cyclosporine powder was given in gelatin capsules. The drug was not absorbed and trials were stopped until absorption from the gastrointestinal tract could be achieved. At this point there was no sensitive chemical or radioimmuno-assay for detecting cyclosporine in blood serum. Stähelin suggested the use of a bioassay that had previously been used at Sandoz for different purposes. The assay used the extent of inhibition of the in vitro proliferation of mitogen stimulated mice lymphocytes (lymphocytes that had been stimulated to divide) to determine the concentration of cyclosporine in blood serum [8]. Later, both radioimmunoassays and chemical assays were developed to test serum concentrations in patients [18].
Now that a way of measuring serum concentration of cyclosporine had been found it was possible to arrange a study into the absorption of cyclosporine. This was carried out in 1977. Initially, three oral preparations were tested on three Sandoz employees, Borel, Stähelin and B. von Graffen. The first preparation, taken by Borel showed strong inhibitory activity in his serum. The preparation was an aqueous solution of water, alcohol and polyoxyethylene(20)-sorbitan-mono-oleate (Tween 80®). The second preparation was a suspension of cyclosporine in olive oil, this showed weak serum activity. The third was a capsule of cyclosporine powder that showed no serum activity. These results lead to the development of both oral and parenteral preparations by the Galenical Department at Sandoz [19].
In 1978 Calne started tests on humans. Seven patients with renal failure were given mismatched cadaver kidney transplants [20]. Initially, cyclosporine was administered on its own and was found to be effective at inhibiting rejection. However, nephrotoxity and hepatotoxicity were observed. A cyclophosphamide was later given along with cyclosporine. One patient died of systemic Aspergillus and Candida infection. Another required an allograft nephrectomy because of pyelonephritis in the graft. The other five patients left hospital with functioning allografts. The results were encouraging but more trials were required.
The next trial undertaken by Calne led to the publication of his report ‘Cyclosporine A initially as the only immunosuppressant in 34 recipients of cadeveric organs: 32 kidneys, 2 pancreases, and 2 livers’ [21]. Although Calne’s publication became one of the most important in the history of clinical transplantation, it contained three pieces of information so troubling that further clinical trials were jeopardised. First was a high incidence of lymphomas. Second none of the kidney recipients had normal graft function. Third, there had been a high patient mortality [6].These results were found to be due to over suppression. When Calne’s patient’s kidney function had been poor he had interpreted this as rejection rather than a toxic effect of cyclosporine and had given prednisolone and a cyclophosphamide derivative which made matters worse. Reduction in cyclosporine dosage allowed continuation of clinical trials.
After further trials it was found that cyclosporine with a combination of steroids gave better control of rejection, preserved renal function, and decreased morbidity. In November 1983, 13 years after its discovery, cyclosporine was approved by the US Food and Drug administration for prevention of transplant rejection.
Borel vs Stähelin
The generally acknowledged discoverer and pioneer of cyclosporine is Jean Francois Borel (Fig. 5). Borel started working at Sandoz in 1970, replacing S. Lazaray as head of the Immunology Department. After obtaining his PhD in 1958 Borel worked mainly in the field of veterinary immunogenetics. In 1965 he moved to the Swiss Research Institute where he studied immunology and inflammation in the Department of Medicine. Then in 1970 he made the move to Sandoz [22].
It was in the immunology department led by Borel that the immunosuppressive activity of cyclosporine was discovered. However Stähelin (Fig. 6) refutes the commonly held view that Borel discovered its activity. Stähelin was in charge of the Pharmacology Department, of which the Immunology Group was a part. ‘The test [immunising mice with sheep erythrocytes] was performed in my [Stähelin’s] laboratory…., except for the titration which was done in Borel’s laboratory……Borel, not being interested in The General Screening Programme, did not see the results until later’ [8].
Borel is also given much of the credit for the further development and promotion of cyclosporine, claiming he pursued its development throughout. Stähelin claims that Borel wanted to drop the development of cyclosporine after the failure to find immunosuppressant activity in dogs dosed with cyclosporine.
It is generally accepted that ‘Borel later was to risk his life…By experimenting on himself before cyclosporine toxicity was known…' [23]. This is in reference to the absorption experiment in which Borel and others participated. Stähelin claims that ‘this was an experiment arranged by B. von Gaffenried.. …The procedure was, of course, a controlled clinical trial, and not a self-administration.’
Inevitably, jealousy can arise when the credit for significant discoveries is attributed to one man. The contribution from others was equally vital in cyclosporine development. With so much mundane and routine work being carried out at Sandoz (1,000 preparations were fed into The General Screening programme each year), it is inevitable that there would be some controversy over the exact details of cyclosporine discovery and development.
NOTE
Further information about the history of cyclosporine, including a range of downloadable manuscripts, can be found at this URL: http://www.sandimmune.ch
Perhaps most credit should go to the scientists K. Saameli, who set up The General screening Programme and S. Lazary, who developed the testing for immunology before Borel had even arrived at Sandoz. The programme ensured that the immunosuppressive properties of any compound entered into it would be found. Luck, however, also played a part. If A. Rüegger had not entered the sample in the programme cyclosporine may have only ever been known as a weak antifungal agent, with no clinical value. However the combination of hard work and good luck bore fruit.
Synthesis
In 1984 synthetic cyclosporine was produced (Fig. 7).It was then possible for cyclosporine to be chemically modified in every possible way. However, none of the derivatives have been found to have greater potency or decreased side effects than cyclosporine itself [24].The two major limitations of cyclosporine therapy today remain its nephrotoxicity and incomplete control of chronic rejection.
Fig. 7. Scanning electron micrograph of cyclosporine crystals in purest form.
In 1996 undergraduate mycology students from Cornell University on a field trip to Ithaca, New York were told to pick up anything that looked interesting. Among the findings was a mysterious fungal fruiting body in an eviscerated beetle grub [25]( Fig. 8). The fungus was later identified as Cordyceps subsessilis an extremely rare fungus that is the sexual state of Tolypocladium inflatum. All the cyclosporine had so far been made from Tolypocladium inflatum cultures without it ever reaching the sexual state. Cordyceps is a large genus that includes around 280 species [26], and may be a good place to start looking, in the estimated 90% of world fungi that have yet to be identified, for a new and improved transplantation drug.
Fig. 8. A beetle larva infested with the sexual stage (Cordyceps subsessilis) of the fungus that produces cyclosporine.
Conclusions
Discovery of cyclosporine led the way to an era of selective lymphocyte inhibition. It enabled the expertise in clinical, technical and immunobiological aspects of transplantation to be put into practice and changed the face of transplantation. Its contribution to autoimmune therapy is less well known but in the long term will probably be of comparable importance.
Cyclosporine did not solve all the problems of transplantation. Today chronic rejection is the main problem. It is poorly understood and there is no treatment for it, although it is thought to have a large immunological component. The majority of transplant patients require long term treatment with high doses of immunosuppressives which increases susceptibility to infection and malignancies.
Thanks to the discovery and development of cyclosporine, patients are alive today years after their operation. Without cyclosporine they would not have survived.
References
1. Borel, J. F. (1986). Ciclosporin and its future. Progress in Allergy, 38: 9-18.
2. Rang, H. P., Dale, M. M. & Ritter, J. M. (1999). Pharmacology. 4th ed. Churchill Livingstone.
3. Kahan, B. D. (1984). Cyclosporine: nursing and paraprofessional aspects. Grune and Stratton.
4. Kahan, B. D. (1999). Cyclosporine: a revolution in transplantation. Transplantation Proceedings, 31: (Suppl 1/2A), 14S-15S.
5. Bach, J. F. (1999). The contribution of cyclosporine A to the understanding and treatment of autoimmune diseases. Transplantation Proceedings, 31: (Suppl 1/2A), 16S-18S.
6. Borel, J. F. & Kis, Z. L.(1991).The discovery and development of cyclosporine (Sandimmune). Transplantation Proceedings,23:1867-1874.
7. Dreyfuss, M., Härri, H., Hofmann, H., Kobel, H., Pache, W. & Tscherter, H. (1976). Cyclosporin A and C. European Journal of Applied Microbiology, 3: 125-133.
9. Wenger, R. M. (1986). Synthesis of Ciclosporin and analogues: structural and conformational requirements for immunosuppressive activity. Progress in Allergy, 38: 46-64.
10. Petcher, T. J., Weber, H. P. & Rüegger, A. (1976). Crystal and molecular structure of an iodo-derivative of the cyclic undecapeptide cyclosporin A. Helvetica Chimica Acta, 59: 1480:157.
11. Wenger, R. (1989). Pharmacology of cyclosporin. Pharmacological Reviews, 41: 243-247.
12. Kessler, H., Loosli, H. R. & Oschkinat, H. (1985). Assignment of the 1H-, 13C, and 15N-NMR spectra of cyclosporin A in CDCl3 and C6D6 by a combination of homo- and heteronuclear two-dimensional techniques. Helvetica Chimica Acta, 68: 661-682.
13. Wenger, R. M. (1984). Total synthesis of ‘cyclosporin A’ and ‘cyclosporin H’, two fungal metabolites isolated from species Tolypocladium inflatum Gams. Helvetica Chimica Acta, 67: 503:515.
14. Quesniaux, V., Tees, R., Schreier, M. H., Wenger, R. M., Donatsch, P. & Van Regenmortel, M. H. V. (1986). Monoclonal antibodies to ciclosporin. Progress in Allergy, 38: 108-122.
15. Borel, J. F. & Wiesinger, D. (1977). Regulatory mechanisms in lymphocyte activation. New York Press,56: 716-721.
16. Borel, J. F., Feurer, C., Gubler, H.U. & Stähelin, H. (1976). Biological effects of cyclosporin A: a new antilymphocytic agent. Agents and Actions, 6: 468-475.
17. Calne, R. Y., White D. J. G., Rolles, K. & Smith, D. P. (1978). Prolonged survival of pig orthotopic heart grafts treated with cyclosporin A. The Lancet, 1182-1185.
18. Schran, H. F., Robinson, W. T., Abisch, E. & Niederberger, W. (1986). Bioanalytical considerations. Progress in Allergy, 38: 73-92.
19. Cavanak, T. & Sucker, H. (1986). Formulation of dosage forms. Progress in Allergy, 38: 65-72.
20. Calne, R. Y., White, D. J., Thiru, S., Evans, D. B., McMaster, P., Dunn, D. C., Craddock, G. N., Pentlow, B. D. & Rolles, K. (1979). Cyclosporin A in patients receiving renal allografts from cadaver donars. Lancet, 2 (8104-5): 1323-1327.
21. Calne, R. Y. ., White, D. J., Thiru, S., Evans, D. B., McMaster, P., Dunn, D. C., Craddock, G. N., Pentlow, B. D. & Rolles, K. (1979). Cyclosporin A initially as the only immunosuppressant in 34 recipients of cadeveric organs: 32 kidneys, 2 pancreases, and 2 livers. Lancet,2: 1327.
22. Stiller, C. R. (1999). Tribute to Jean Francois Borel: A gentleman and a scholar. Transplantation Proceedings, 31: (Suppl 1/2A), 3S-8S.
23. Bernard, J. (1986). Ciclosporin: foreward. Progress in Allergy, 38: 1-8.
24. Wenger, R. M. (1982). Cyclosporin A. Biomedical Journal, 3: 19-31