
4.3 The origin of drugs in current use: the ergot alkaloids story (contributed by Ziad Madlom, 2002)
Abstract
Ergot is a fungal infection that has infected rye and other plants since farming began. One of the constituents of ergot, the ergot alkaloids, were found to have useful medicinal properties. Ergot was known to cause gangrene in the limbs of those who had ingested infected bread. But later its first medicinal property as a powerful oxytocic (facilitating childbirth) was discovered, and more recently its derivatives have been used in the treatments of migraine. It was the alkaloids of ergot that were found to be the active components in their pharmacological actions, starting with the first pure alkaloid to be isolated, ergotamine. This review covers the history of ergot and the ergot alkaloids and tries to show how ergot went from being just an infection of grass to its alkaloids being the active component in many drugs, especially those in the treatment of migraine. The mechanism of action of the ergot alkaloids is also explained.
Introduction
Throughout history man has turned to nature for medicines. Microorganisms, such as fungi, offer a huge source of pharmaceutically useful molecules. For the fungi the relatively recent discovery of penicillin marked a new era. Of the top 20 selling prescription medicines in 1995, six of these contained compounds derived from fungi [1].
 |
| Fig. 1. Ergots (the blackened claw-like objects) of Claviceps purpurea on wild oat, Avena fatua. Photograph of the dried seed head by David Moore. |
Ergot is the dried sclerotium of the fungus Claviceps purpurea, which can arise in the ovary of the rye Secale cereale. These sclerotia can replace one or more of the kernels in a mature grain head with a hard, dark coloured, horn-like mass (Fig. 1). Although ergot most readily attacks rye it can also infect other wild and cultivated cereals (barley, wheat, oats), and other grasses. These sclerotia contain the fungal toxins and are harvested along with the rest of the grain so that the ergots contaminate the food chain. Widespread contamination by ergot, known as ergotism, occurred frequently during the Middle Ages. Eating rye bread contaminated with C. purpurea resulted in gangrene of limbs, disruption in functions of the CNS and even death. Ergot does however have its uses in medicine, with midwives as far back as 1582 using it to reduce haemorrhage after childbirth [2]. Ergotamine, an alkaloid of ergot, has been found to be useful in the treatment of migraine headaches.
History
It is possible that ergot infected grasses produced in the first agricultural settlements of Mesopotamia around 9000 BC, but ergot is thought to have first been mentioned around 600 BC by the Assyrians [2]. The Roman historian Lucretius (98-55 BC) referred to ergotism as ‘Ignis sacer’, meaning Holy Fire, which was the name given to ergotism during the Middle Ages, and it was during these times that ergotism occurred frequently.
The word ergot is actually derived from an old French word argot, meaning the cock’s spur. The violet or black sclerotia formed by ergot consist of a mass of hyphae, and may be two to ten times the length of a normal kernel. When bread was prepared from grain contaminated by the black spurs, epidemics of ergotism occurred. Poor people mainly ate rye bread, especially during famines, when even the spurs were collected because of hunger. The first mention of gangrenous ergotism was in Germany in 857 AD, and the first epidemic of convulsive ergotism occurred in 945 in Paris. These are the two distinct types of ergotism, with the gangrenous type seen mostly in France and the convulsive one in Germany. The name Holy Fire or St. Anthony’s Fire given to ergot epidemics during the Middle Ages referred to the burning sensation in the limbs experienced by sufferers.
Ergot was probably first used in medicine as an oxytocic drug. In 1582 Adam Loncier in Germany made the first note of ergot stimulating uterine contractions of labour by administering three sclerotia [2]. It was the most effective drug for this purpose at the time resulting in a rapid and sudden termination of labour, with a delivery time lasting less than three hours. But ergot was eventually deemed unsuitable for this purpose as the dosage could not be given accurately due to large variations in the active ingredients. Ergot also caused severe adverse effects, such as violent nausea and vomiting. And it was in 1822 when Hosack from New York stated that many stillbirths were due to uterine rupture resulting in maternal death, and the use of ergot as an oxytocic was virtually abandoned by the end of the 19th century [2]. It was only during the 20th century that ergot was shown to be useful in the treatment of attacks of migraine. This would mainly involve the alkaloid of ergot, ergotamine.
Life cycle
As already mentioned, ergot is derived from the fungus Claviceps purpurea and is formed in the body of the sclerotium of the fungus that grows mainly in the seed of the rye plant and various other cereal grasses. There are 32 recognized species of ergot. The sclerotia of the fungus will remain dormant through periods of winter or drought and will only germinate under favourable, moist conditions to give rise to small club-shaped fruiting bodies. The heads of these fruiting bodies are shiny and spherical and produce long, filamentous ascospores, which are the sexual spores of the fungus.
The ascospores are ejected from the fruit body of the fungus and are blown by the wind. If they land on a healthy stigma of a grass flower or rye plant, they germinate and their hyphae enter, as do pollen tubes, into the plant's ovary and begin to form a fine mycelial network. Soon after infection, a special structure called the sphacelium develops between the ovary and the base of the floral cavity. The sphacelium produces hundreds of thousands of conidia. These conidia are the asexual spores of the fungus. They are embedded in a sugary matrix called honeydew, a yellowish saccharine secretion produced by the sphacelia, which will reduce Fehling’s solution. The honeydew is very sticky and its strong, fetid smell attracts insects to the plant to feed on these sugars. When the insects move on to another plant, conidia stuck to their legs and mouthparts infect the healthy ovaries of other plants. After several weeks, the sclerotium develops between the sphacelium and the flower base, bringing us back to the beginning of the cycle and completing the life cycle.
Composition of ergot
The ergot sclerotium contains up to 30-40% of fatty oils and up to 2% of alkaloids [3]. The other components of the sclerotium are free amino acids, ergosterin, choline, acetylcholine, betaine, ergothionine, uracil, guanidine, free aromatic and heterocyclic amines (tyramine, histamine, agmatine) and alkylamines (the natural representatives of which were originally found in ergot). The outer shell of sclerotium contains acid pigments belonging to anthraquinolinic acid derivatives, including orange- red (endocrinin, clavorubin) and light yellow (ergochromes, ergochrysins). These pigments form part of the sclerotium shell giving rise to the greyish-brown-violet colour. Albino ergot strains also exist which are incapable of producing pigments. Naturally growing Claviceps purpurea species has several geographic types differing in both qualitive and quantitative composition of alkaloids. So ergot strains are specially selected that are capable of producing predominantly a single alkaloid or a certain group of alkaloids: ergotamine, ergotoxine, ergocristine, etc.
The ergot alkaloids have a high biological activity and a broad spectrum of pharmacological effects, hence they are of considerable importance to medicine. They have adrenoblocking, antiserotonin and dopaminomimetic properties. Ergot alkaloids have a therapeutic effect on some forms of migraine, post-partum haemorrhages, mastopathy, and a sedative effect on the central nervous system [6]. These compounds are now obtained both by methods of artificial parasitic cultivation on rye and by techniques using in vitro culture .
Naturally occurring ergot consists of two types of alkaloids. The first, the clavine-type alkaloids, are derivatives of 6,8-dimethylergoline. The second type comprises the lysergic acid derivatives, which are peptide alkaloids. All ergot alkaloids can be considered as derivatives of the tetracyclic compound 6-methylergoline [4]. It is the lysergic acid derivatives, or peptide alkaloids, that are the pharmacologically active alkaloids. Each active alkaloid occurs with an inactive isomer involving isolysergic acid. These alkaloids have been studied over many years and were not easy to characterize. Since its isolation in 1906 (by Barger and Carr and independently by Kraft), ergotoxine had been accepted as a pure substance, and in the form of ergotoxine ethanosulphate was formerly used as a standard. It was shown to be a mixture of the three alkaloids ergocristine, ergocornine and ergocryptine [5].
Six pairs of alkaloids predominate in the sclerotium and fall into the water-soluble ergometrine group or the water-insoluble ergotamine and ergotoxine groups (Table 1). Alkaloids of groups 2 and 3 are polypeptides in which lysergic acid or isolysergic acid is linked to other amino acids. In the ergometrine alkaloids lysergic acid or its isomer is linked to an amino alcohol. Ergometrine was isolated in 1932 by Dudley & Moir [4], and was synthesized by Stoll & Hoffmann in 1943 [5].
| Table 1. Alkaloids of the ergot sclerotium | |||
| Group | Alkaloid | Formula | Discovered |
| 1.Ergometrine group | Ergotmetrine
Ergotmetrinine | C19H22O2N3
| Dudley & Moir (1935) |
| Ergotamine | C33H35O5N5
| Spiro & Stoll (1920) | |
| 2.Ergotamine group | Ergotaminine | ||
| Ergosine | C30H37O5N5
| Smith & Timmis (1937) | |
| Ergosinine | |||
| Ergocristine | C35H39O5N5
| Stoll & Burckhardt (1937) | |
| 3.Ergotoxine group | Ergocristinine | ||
| Ergocryptine | C32H41O5N5 | Stoll & Hofmann (1938,1943) | |
| Ergocryptinine | |||
| Ergocornine | C31H39O5N5 | ||
| Ergocorninine | |||
Alkaloid production in artificial cultures
Ergot alkaloids can be produced in submerged fermentation by Claviceps or Penicillium species, which are used for their industrial production. Initial work in Japan showed that submerged cultures did not produce the typical alkaloids associated with the sclerotium but instead produced a series of new non-peptide bases (clavines) which did not posses any significant pharmacological action. Attempts were made by many workers to influence alkaloid production by modification of the culture medium and the fungus strain. The first pure ergot alkaloid, ergotamine, was obtained by Stoll in 1920. Moir reported the discovery of the “water soluble uterotonic principle of ergot” in 1932 [8]. This was subsequently determined to be ergonovine (also called ergometrine). As a result of successful experiments in 1960 [5], the commercial manufacture of simple lysergic acid derivatives by fermentative growth of a strain of Claviceps paspali became feasible.
The final fermentation broth contains a complex mixture of alkaloids, salts, polysaccharides, fats, solids, etc.; and organic solvent extraction is commonly used for the separation of ergot alkaloids from the broth. To avoid the formation of emulsions during the extraction, a separation process taking advantage of solid-liquid adsorption of alkaloids on activated carbon bentonite and other silicate sorbents was developed in 1986 [7]. In 1988 selective adsorption of polycarboxylic ester resin XAD-7 to isolate alkaloids from plant cell cultures was used [7]. In 1990 the Kawaken Fine Chemicals Company submitted two patents for isolation and purification of ergot alkaloids from Claviceps species fermentation broth by adsorption on porous, synthetic polymer, ion-exchanging adsorbents such as styrene divinylbenzene copolymer or phenolic resin. The fermentation broth was passed through a packed bed column and subsequently washed by water and eluted by methanol. Methanol was removed under reduced pressure. The residue was extracted with ethyl acetate and dried with a yield of 68% and purity of 94%. In more recent studies, the simultaneous uptake of agroclavine, elymoclavine, chanoclavine and chanoclavine aldehyde by inorganic sorbents has been investigated (Votruba & Flieger, ref. 7).
Chemistry
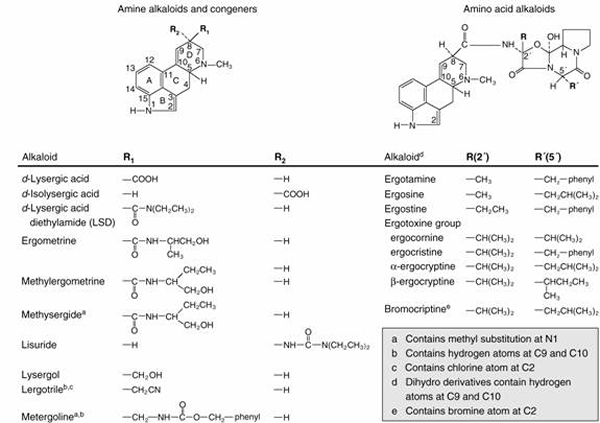 |
| Fig. 2. An abbreviated catalogue of naturally occurring ergot alkaloids. |
The naturally occurring alkaloids contain a substituent in the β configuration at position 8 and a double bond in ring D (Fig. 2). The natural alkaloids of therapeutic interest are amide derivatives of δ-lysergic acid; these compounds contain a double bond between C9 and C10 and thus belong to the family of 9-ergolene compounds. Many alkaloids, containing either a methyl or a hydroxymethyl group at position 8, are present in ergot in small quantities. These are the clavine alkaloids and consist mainly of both 9-ergolenes (e.g. lysergol) and 8-ergolenes (e.g. elymoclavine, the 8-ergolene isomer of lysergol).
Optical isomerism exhibited by the ergot alkaloids is due to the two asymmetrical carbon atoms (position 5 and 8) in the lysergic part of the molecule. Derivatives of l-lysergic acid (the epimer at position 5) and of δ-isolysergic acid (the epimer at position 8) display relatively little biological activity. Upon hydrolysis, ergonovine and its derivatives yield lysergic acid and an amine; consequently they are called amine alkaloids. The alkaloids of higher molecular weight yield lysergic acid, ammonia, pyruvic acid (or a derivative of), proline, and one other amino acid (either phenylaline, leucine, isoleucine, or valine) and so are known as amino acid alkaloids.
Numerous semisynthetic derivatives of the ergot alkaloids have been prepared, and several are of therapeutic interest. The earliest derivatives were prepared by the catalytic hydrogenation of the natural alkaloids, yielding a series of compounds that are saturated in ring D of lysergic acid. These have been designated dihydroergotamine, dihydroergocristine, and so on, and possess different pharmacological properties than do the parent alkaloids. Another ergopeptine derivative is bromocriptine (2-bromo-α-ergocryptine). It is also possible to prepare different amides of lysergic acid. Two products of this series, lysergic acid diethylamide (LSD) and lysergic acid hydroxybutylamide (methylergonovine) are of pharmacological interest. Methylation of the indole nitrogen of the latter compound yields 1-methylmethylergonovine (methysergide). A large number of related compounds that are not derivatives of lysergic acid have also been prepared. These include lisuride, lergotrile (2-chloro-6-methyl-8β-cyanomethyl-ergoline), and metergoline (1,6-dimethyl-8β-carbobenzoxyaminomethyl-ergoline).
Pharmacological properties
In general, the effects of all the ergot alkaloids appear to results from their actions as partial agonists or antagonists at adrenergic, dopaminergic, and tryptaminergic receptors. The spectrum of effects depends on the agent, dosage, species, tissue, and experimental or physiological conditions. However, some of the actions of the ergot alkaloids are not entirely compatible with this view: (1) while agonistic effects are generally apparent only at concentrations that are lower than those required to observe antagonism, this is not always the case (e.g. the action of methysergide on cerebral blood vessels); (2) the effects of full agonists (e.g. norepinephrine) are usually augmented by low concentrations of ergot alkaloids, even those with weak efficacy as partial agonists (e.g. the action ergonovine on arterioles); and (3) the contractile responses to other agents, such as acetylcholine or angiotensin, are sometimes also augmented by adrenergic or tryptaminergic blocking agents. These and other observations emphasize the importance of the physiological or pathophysiological state in determining the spectrum and intensity of effects produced in animals or patients.
Apart from the stereochemical considerations mentioned above, few rules governing structure-activity relationships have emerged. In general, small amide derivatives of lysergic acid are potent and relatively selective antagonists of 5-HT, while the amino acid alkaloids are usually less selective and show similar affinities as blocking agents at α-adrenergic and tryptaminergic receptors. Dihydrogenated derivatives usually have fewer and less intense agonistic actions than do parent alkaloids. Finally, insertion of a methyl group at position 1 usually results in compounds with less affinity for receptors for catecholamines and with more selective ability to block tryptaminergic receptors.
All of the natural alkaloids of ergot significantly increase the motor activity of the uterus. After small doses contractions are increased in force or frequency, or both, but are followed by a normal degree of relaxation. As the dose is increased, contractions become more forceful and prolonged, resting tonus is markedly increased, and sustained contracture can result. Although this characteristic precludes their use for induction or facilitation of labour, it is quite compatible with their use postpartum or after abortion to control bleeding and maintain uterine contraction. The gravid uterus is very sensitive, and small doses of ergot alkaloids can be given immediately postpartum to obtain a marked uterine response, usually without significant side effects.
Effects on the uterus
Although all natural ergot alkaloids have qualitatively the same effect on the uterus, ergonovine is most active and also less toxic than ergotamine. For these reasons ergonovine and its semi-synthetic derivative methylergonovine have replaced other ergot preparations as uterine-stimulating agents in obstetrics. Methylergonovine differs little from ergonovine in its uterine actions. The dihydrogenated alkaloids do not have the uterine-stimulating properties of the parent alkaloids when tested in experimental animals; however they are capable of exerting a marked uterine-stimulating action on the pregnant human uterus at term. Ergonovine and methylergonovine are rapidly and virtually completely absorbed after oral administration and reach peak concentrations in plasma within 60 to 90 minutes that are ten times those achieved with an equivalent dose of ergotamine. A uterotonic effect can be observed within 10 minutes after oral administration of 0.2 mg of ergonovine to women postpartum. Judging from the relative duration of action, ergonovine is metabolised and/or eliminated more rapidly than ergotamine. The half-life of methylergonovine in plasma ranges between 0.5 and 2 hours [8].
Effects on the cardiovascular system
The natural amino acid alkaloids, particularly ergotamine, constrict both arteries and veins. While dihydroergotamine retains appreciable vasoconstrictor activity, it is far more effective on capacitance than on resistance vessels. This property is the basis for investigation of its usefulness in the treatment of postural hypotension. The dihydrogenated derivatives of the ergotoxine group are considerably less active and usually produce hypotension because of effects on the CNS. In doses used in the treatment of migraine, the rectal administration of ergotamine produces little change in blood pressure but does cause a slowly progressing increase in peripheral vascular resistance that persists for up to 24 hours [8]. This could reflect constriction of arteries mediated by stimulation of trypaminergic receptors. Dihydroergotamine has relatively little capacity to produce such effects in human beings.
At the higher plasma concentrations achieved by intravenous administration, both ergotamine and dihydroergotamine cause a rapid increase in blood pressure that dissipates in a few hours. This is thought to reflect action on arterioles, which in rats results from stimulation of α2-adrenergic receptors. With the notable exception of the cerebrum, the prolonged increase in vascular resistance is accompanied by decreased blood flow in various organs. This results in part from reduced flow through non-nutritive arteriovenous anastomoses. While less potent than ergotamine, the amine alkaloids can also raise blood pressure slightly and decrease blood flow in the extremities when administered in therapeutic doses. The intensity of pressor effects is greater when the blood pressure is raised. Ergot alkaloids that produce peripheral vasoconstriction can also damage the capillary endothelium. The mechanism of this toxic action is poorly understood. Vascular stasis, thrombosis, and gangrene are prominent features of ergot poisoning. The natural tendency of these alkaloids to cause gangrene appears to parallel their vasoconstrictor activity.
Pharmacological properties related to treatment of migraine
Ergot derivatives were first found to be effective anti-migraine agents in the 1920s, and they continue to be a major class of therapeutic agents for the acute relief of moderate or severe migraine. However, ergot alkaloids are non-selective pharmacological agents in that they interact with numerous neurotransmitter receptors, including all known 5-HT1 and 5-HT2 receptors, as well as adrenergic and dopaminergic receptors. As shown in Table 2, the ergot alkaloid dihydroergotamine (DHE) can compete for radioligands for binding to a variety of receptor subpopulations.
DHE is potent at all known 5-HT1 receptors as well as at a number of other biogenic amine receptors, such as 5-HT2A, 5-HT2B, D2 dopamine, and α1- and α2-adrenergic receptors. The multiple pharmacological effects of ergot alkaloids have complicated the determination of their precise mechanism of action in the acute treatment of migraine. These findings should be compared with those for the selective 5-HT receptor agonist, sumatriptan, which has its highest affinity for 5-HT1D and 5-HT1B receptors, with lower affinity for 5-HT1A and 5-HT1E receptors.
| Table 2. Ergot alkaloid reactivity with neurotransmitter receptors | |||
|
Receptor | Ki Values (nM) | ||
| Dihydroergotamine | Sumatriptan | ||
| Serotonergic | |||
| 5-HT1D | 0.55 | 6.7 | |
| 5-HT1A | 0.83 | 120 | |
| 5-HT1B | 6.2 | 35 | |
| 5-HT1E | 8.8 | 920 | |
| 5-HT2C | 39 | >10,000 | |
| 5-HT2A | 78 | >10,000 | |
| 5-HT3 | >10,000 | >10,000 | |
| Adrenergic | |||
| a1 | 6.6 | >10,000 | |
| a2 | 3.4 | >10,000 | |
| b | 960 | >10,000 | |
| Dopaminergic | |||
| D1 | 700 | >10,000 | |
| D2 | 98 | >10,000 | |
| Other sites | |||
| Muscarinic | >10,000 | >10,000 | |
| Benzodiazepine | >10,000 | >10,000 | |
Absorption and excretion
The oral administration of ergotamine by itself results in undetectable systemic drug concentrations because of extensive first-pass metabolism. Bioavailability after sublingual administration also is poor and is often inadequate for therapeutic purposes. Although the con-current administration of caffeine improves both the rate and extent of absorption, the bioavailability of ergotamine is probably still less than 1%. The bioavailability after administration of rectal suppositories is greater, and maximal plasma concentrations of ergotamine of over 400 pg/ml can be achieved following a 2-mg dose, compared to peak plasma concentrations of approximately 20pg/ml in plasma 70 minutes after a 2-mg dose taken orally.
Ergotamine is metabolised in the liver by largely undefined pathways, and 90% of the metabolites are excreted in the bile. Only traces of unmetabolized drug can be found in urine and faeces. Ergotamine produces vasoconstriction that lasts for 24 hours or longer, despite a plasma half-life of about 2 hours. Dihydroergotamine is much less completely absorbed and is eliminated more rapidly than ergotamine, presumably due to its rapid hepatic clearance [8].
Ergot derivatives and Parkinson’s disease
Recent studies involving ergot derivatives in the treatment of Parkinson’s disease have investigated their effects on dopamine receptors [9-10]. Most dopamine agonists employed at present are ergot derivatives. The similarity of the ergoline ring structure to the endogenous monoamines explains the action of these compounds on dopaminergic, serotonergic and adrenergic receptors. Bromocriptine, lisuride and pergolide are the three oral ergot dopamine agonists presently available [9].
Bromocriptine, which has potent D2 agonist properties and a weak D1 receptor antagonistic effect, was the first ergoline to be successfully used in Parkinson’s disease. The synthetic ergoline derivative pergolide is a long acting agonist at D2 dopamine receptors and has a mild D1 receptor agonistic effect. Both bromocriptine and pergolide are effective in relieving the symptoms of Parkinson’s disease and can achieve a reduction of on-off fluctuations. Comparative studies of both drugs have shown that they have similar efficacy and adverse effects [9]. The efficacy of a long acting, slow release formulation of bromocriptine has been shown to be equivalent to standard treatment with bromocriptine, but patients needed fewer doses daily and had fewer adverse effects.
Another ergot derivative, lisuride, stimulates postsynaptic striatal D2 receptors and is a mild D1 receptor agonist. The antiparkinsonian efficacy of this drug is equivalent to that of bromocriptine and pergolide. Lisuride is highly water-soluble and can therefore be used for parenteral therapy via ambulatory infusion pumps. This compound has been shown to be very effective in controlling motor fluctuations in Parkinson’s disease when administered by continuous infusion. However, long-term studies of lisuride have shown that its parenteral use is complicated by a high incidence of psychiatric adverse effects, possibly because of its serotonergic properties [9].
Conclusion
The isolation of the ergot alkaloids from ergot revolutionized the treatment of attacks of migraine, and also provided an alternative oxytocic. LSD however is probably the most famous ergot derivative, even though it has the least use in medicine. The long history of ergot and ergotism means that the use of ergot alkaloids in drugs comes only after centuries of infection of grass and intoxication of the ancients and mainly the poor during the Middle Ages. Although ergot derivatives have been superseded in the treatment of migraine by drugs such as sumatriptan, their influence means their place in history is assured. But other applications of the ergot alkaloids, such as their use to treat the symptoms of Parkinson’s disease, could mean that they provide the active components of many mainstream drugs for years to come.
References
1. Langley, D. (1998). Exploiting the fungi: novel leads to new medicines. Mycologist, 11: 165-166. DOI: http://dx.doi.org/10.1016/S0269-915X(97)80094-8.
2. van Dongen, P.W. J. & de Groot, A.N.J.A. (1995). History of ergot alkaloids from ergotism to ergometrine. European Journal of Obstetrics & Gynaecology and Reproductive Biology, 60: 109-116. DOI: http://dx.doi.org/10.1016/0028-2243(95)02104-Z.
3. Komarova, E.L. & Tolkachev, O.N. (2001). The chemistry of peptide ergot alkaloids: Part 1. Classification and chemistry of ergot peptides. Pharmaceutical Chemistry Journal, 35: 504-506. DOI: http://dx.doi.org/10.1023/A:1014050926916.
4. de Groot, N.J.A., van Dongen, P.W.J., Vree, T.B., Hekster, Y. A. & van Roosmalen, J. (1998). Ergot alkaloids - current status and review of clinical pharmacology and therapeutic use compared with other oxytocics in obstetrics and gynaecology. Drugs, 56: 523-535. URL: http://www.ncbi.nlm.nih.gov/pubmed/9806101.
5. Evans, W.C. (1996). Pharmacognosy. 14th edition. WB Saunders Company Ltd, London.
6. Boichenko, L.V., Boichenko, D.M., Vinokurova, N.G., Reshetilova, T.A. & Arinbasarov, M.U. (2001). Screening for ergot alkaloid producers among microscopic fungi by means of the polymerase chain reaction. Microbiology, 70: 306-307. DOI: http://dx.doi.org/10.1023/A:1010403427611.
7. Votruba, V. & Flieger, M. (2000). Separation of ergot alkaloids by adsorption on silicates. Biotechnology Letters, 22: 1281-1285. DOI: http://dx.doi.org/10.1023/A:1005693200572.
8. Hardman, J.G. & Limbird, L.E. (eds.) (1996). The Pharmacological Basis of Therapeutics, 9th edition. McGraw-Hill
9. Lange, K.W. (1998). Clinical pharmacology of dopamine agonists in Parkinson's disease. Drugs & Aging, 13: 381-389. PDF
10. Thompson, F., Muir, A., Stirton, J., Macphee, G. & Hudson, S. (2001). Parkinson’s disease. The Pharmaceutical Journal, 267: 600-612.
Updated December 7, 2016